DADA2解説1:Install、Quality check、Filter and Trim
微生物分野ではよく使われるようになってきた、ASVによるクラスタリングをしてくれるDADA2。
一般的に、いわゆる次世代シーケンサーを用いた Amplicon sequencing によって得られたデータは、得られた塩基配列情報を 95 ~ 98 % 類似していたら、同じ種であろうと推測し、 OTU という仮の種名みたいなもの / 分類群の単位を与えて分析してきた。
しかし、Microbiome の場合は、1塩基違うだけでも全く別の種、機能的に異なることがある。
そこで、一塩基の違いが strain / 種特異的なものか、PCR errorによるものかを判別し、read をクラスタリングしたものに ASV : Amplicon sequence variant という単位を与えて分析する。
以上が、ASV が微生物分野で使われるようになってきた理由だと思います。
いつか、原著をちゃんと読んで、このブログ内でまとめられたらと思います。
それはそれとして、肝心の DADA2 の使い方というのが、英語でしか書かれていなく、Bioinfomatics初心者にはハードルが高いように思ったので、すこしずつ DADA2 の Function の使い方を紹介していけたらと思います。
では、DADA2 のチュートリアルに沿って話していきます。
DADA2 Pipeline Tutorial (1.16)
DADA2 のインストール
DADA2 は R で動かすことができます。
package のインストールにはいろいろな方法があるようです(
https://benjjneb.github.io/dada2/dada-installation.html)。
今後のことも考えると以下のがおすすめです。
単純に、devtoolsを介したpackageのインストールが他の場面でもありえるからです。
install.packages("devtools")
library("devtools")
devtools::install_github("benjjneb/dada2", ref="v1.16")インストールが終わったら、Test dataを落としてみてもいいかもしれないです。
170MBていどなので、そんなに邪魔にならないとは思います。
また、ここからはTest data使って話していきます。
Test data at https://benjjneb.github.io/dada2/tutorial.html
https://mothur.s3.us-east-2.amazonaws.com/wiki/miseqsopdata.zip
read のクオリティチェック
DADA2 の workflow は
1. read のクオリティチェック
2. Low quality read のフィルタリング
3. Error rate の計算
4. ASV の推定
です。
なので、サンプルごとに fastq を分けておく (demultiplexをする) 必要があります。
参考:Ushio's blog: DADA2 と Claident を用いた short-read amplicon sequence のデータ解析(こちらのサイトでも、分かりやすくDADA2の使い方について解説しています)
ここでは、demultiplex されたこと前提で話を進めます。
どうやら、DADA2 は fastq.gz ファイルを直接読み込んで動作するようです。
なので、最初に fastq.gz ファイルをまとめて入れたフォルダを指定、path を通しておきます。
path <- "~/MiSeq_SOP" # fastq.gz が入ったフォルダの名前を入れる head( list.files(path) ) # list.files() で指定したフォルダの中身を確認。head() で最初の6個だけ表示
# Mac または linux で、R console または Terminal からRで作業していたら、フォルダをコンソール画面にドラッグするだけで、絶対パスを表示してくれます。

ちゃんと指定ができていたら、フォルダの中身がずらっと表示されるはずです。
では、各サンプルの quality をプロットします。その結果を見て、どういうパラメーターでフィルタリングするかを決めていきます。
fnFs <- sort(list.files(path,
pattern="_R1_001.fastq",
full.names = TRUE) )# フォワード側の fastq ファイルの名前のリストを取り出します。ここでは、R1がフォワードと同義なので、list.files関数の pattern オプションで "_R1_001.fastq" という文字列が入ったファイルだけ抜き出しています。なので、pattern の中身は必要に応じて変えてください
fnRs <- sort( list.files(path,
pattern="_R2_001.fastq",
full.names = TRUE))# Paired-end の場合は、reverse 側も取り出します。
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)
# サンプルID用の文字列を作ります。
この中身は、Rに慣れてないとややこしいので、分解して説明します。
上で取り出したfnFsは、絶対パス、フォルダの階層を含めた名前なっていますので、そこからファイル名だけをbasename(fnFs) で、取り出します
# 例 > fnFs="path1/path2/F3D0_S188_L001_R1_001.fastq" > basename(fnFs) [1] "F3D0_S188_L001_R1_001.fastq"
次に、strsplit(basename(fnFs), "_") で、文字列を "_"で切り分けます。
# 例 > fnFs="path1/path2/F3D0_S188_L001_R1_001.fastq" > basename(fnFs) [1] "F3D0_S188_L001_R1_001.fastq" > strsplit(basename(fnFs), "_") [[1]] [1] "F3D0" "S188" "L001" "R1" "001.fastq"
ただ、strsplitの結果は list クラスで返してきますので、list に働きかける sapply () で取り出していきます。
# 例
> fnFs="path1/path2/F3D0_S188_L001_R1_001.fastq"
> basename(fnFs)
[1] "F3D0_S188_L001_R1_001.fastq"
> strsplit(basename(fnFs), "_")
[[1]]
[1] "F3D0" "S188" "L001" "R1" "001.fastq"
> sapply( strsplit(basename(fnFs), "_"), # List オブジェクト
`[`, # 関数。なんで ` [ ` で動くのかはわからない...ベクトルをとってきているのかな?
1 # 各リストの一個目の要素を取り出す。ここを2にしたら "S188"が返る
)
[1] "F3D0"ややこしくなりましたが、ここの流れで固有のサンプル名が取り出せるとおもいます。自分で変えるべきとこはstrsplitの文字区切りのところと、区切った文字列の何番目を取り出すかです。
では、fastq.gz ファイルを指定して、クオリティを図示します。
plotQualityProfile(fnFs[1:2])
# フォワード側のファイルの1~2個目を図示します。
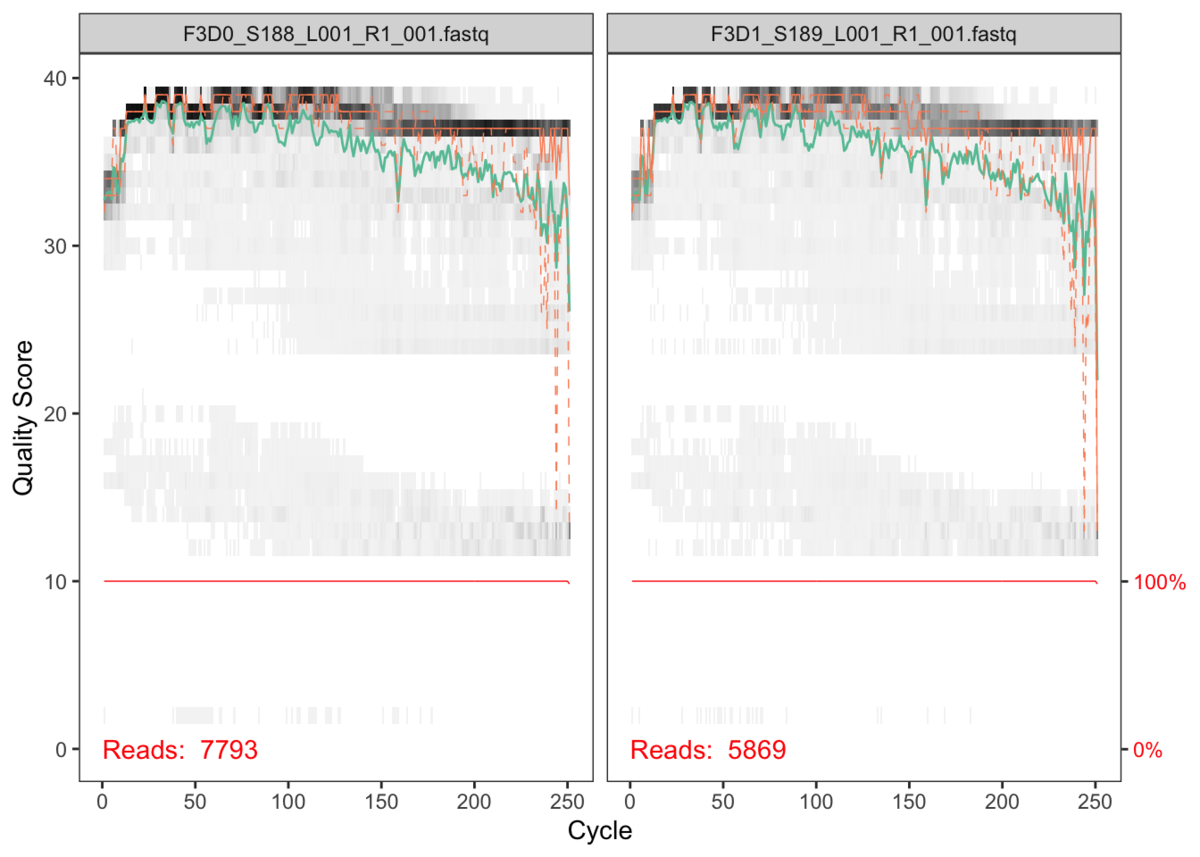
以下のような図が表示されます。
DADA2の公式サイトから画像をお借りしました。
{kind=link}
グレーのヒートマップで示されている部分が、あるサンプル内の read の頻度です。黒い方が頻度が高いです。
緑の折れ線が、全リードの Quality score の平均。オレンジが Quality score の 25 / 75 パーセンタイルです。
赤い線は...よくわからないです...。おそらく、それぞれの read の長さが揃っていたら 100%を示すのかなと思います。
基本的に、Illumina sequencer のデータではQuality score が 30 以上だったら良いです。
考え方は、クオリティスコア | シークエンサーが出力するクオリティスコアとシーケンシングエラー率から引用すると、
Quality score = -10 log10 エラー率
式変換すると
エラー率= 10 ^ -Quality score/10
と表せます。
ここから、Quality score が 30 のときは、シーケンサーが塩基を間違えて読む確率は 0.001 %
つまり、塩基の信頼度は 99.9 % といえるのです。
プロット画像から判断すると、Quality score が 30 を切る cycle が、だいたい 240 cycle 以降なので、それ以降の Cycle は捨てた方がいいということがわかります。
ここまでのコードをまとめると
path <- "~/MiSeq_SOP" # fastq.gz が入ったフォルダの名前を入れる。ユーザーによって変更する箇所
head( list.files(path) ) # list.files() で指定したフォルダの中身を確認。head() で最初の6個だけ表示
# フォワード側のファイル、リバース側のファイルをそれぞれ取り出す
fnFs <- sort(list.files(path,
pattern="_R1_001.fastq", #ユーザーによって変更する箇所
full.names = TRUE) )
fnRs <- sort( list.files(path,
pattern="_R2_001.fastq", #ユーザーによって変更する箇所
full.names = TRUE))
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)#ユーザーによって変更する箇所
# Quality の確認
plotQualityProfile(fnFs[1:2])
plotQualityProfile(fnRs[1:2])
Filter and trim
上記で、Quality score をチェックしたら、それを元に Quality score が低い read を削っていきます。
初めに、Output 先を指定するための文字列を作成します。
filtFs <- file.path(path, "filtered", paste0(sample.names, "_F_filt.fastq.gz")) filtRs <- file.path(path, "filtered", paste0(sample.names, "_R_filt.fastq.gz")) names(filtFs) <- sample.names names(filtRs) <- sample.names
# file.path() が、入れた文字列を' \ ' で区切って、一つの文字列に変換してくれるpasteみたいな関数。paste0() は paste() のsep=''にしたもの。
names()で、作った文字列ベクトルに名前をつけてます。
では、メインの filterAndTrim 関数についてみていきます。
optionはこんだけ指定できます。
中の数字は default の数字です。
filterAndTrim( fwd, # Forward 側のfastq ファイルの絶対パスの羅列 filt, # Forward 側のフィルタリングした結果のアウトプット先のファイル名。アウトプットファイルは勝手に作ってくれます rev = NULL, # Reverse 側のfastq ファイルの絶対パスの羅列 filt.rev = NULL, # Reverse 側のフィルタリングした結果のアウトプット先のファイル名 compress = TRUE, # .gz で圧縮するかどうか。Trueで圧縮します truncQ = 2, # よくわからないです...。このあたりよりQuality scoreが低い read を捨てるのですが、ここでいうQuality scoreがよくわからない。1サイクル目なのか、平均のQuality scoreなのか...。だとしても、defaultがこんなにゆるくていいのか...。 truncLen = 0, # 何サイクル目以降を切り捨てるか。例えば、240 サイクル以降を捨てるときは、ここで240 と指定します。また、これより短い read は自動的に捨てられます。 trimLeft = 0, # 1サイクル目から何サイクル目まで捨てるかを指定できます。基本0でいいと思います。 trimRight = 0, # 最後のサイクルから何サイクル目まで捨てるかを指定できます。truncLenと似たOptionで、trimRightが行われた後にtruncLenが働きます。 maxLen = Inf, # TrimmingとTrancationを行なった後に、指定した値よりも大きい長さのreadを捨てます。 minLen = 20, # TrimmingとTrancationを行なった後に、指定した値よりも短い長さのreadを捨てます。 maxN = 0, # Trancationを行なった後に、指定した値よりもNが多いreadを切り捨てます。DADA2はNを許さないので、無条件で0にした方がいいです。 minQ = 0, # Trancationを行なった後に、指定した値より低いQuality scoreを含む read を捨てます。 maxEE = Inf, # Trancationを行なった後に、指定した値より大きいexpected errorsを示した read を捨てます。expected errors:EE は sum(10^(-Q/10))によって計算されます。つまり、EE = 各 read の合計のエラー率です。 rm.phix = TRUE, # 塩基の多様性を上げるために入れたphiXの配列を除きます。 rm.lowcomplex = 0, # 塩基配列が単調な read を捨てます。これ以降はあまり考えないの省略します。 orient.fwd = NULL, matchIDs = FALSE, id.sep = "\\s", id.field = NULL, multithread = FALSE, n = 1e+05, OMP = TRUE, qualityType = "Auto", verbose = FALSE )
調べてから気づいたのが、Quality score を使ってフィルタリングできる箇所が、maxEEとtruncQだけでした。
なので、最近は別のプログラムでフィルタリングした方がいいのではという気にもなっています。例えば、Claident や fastp というプログラムでは、最後のサイクルから、例えば、5塩基の平均Quality scoreが30以下になる cycle を捨てるといったフィルタリングの方法があります。
そういった考えでTrancationを行う方が、目で見て長さを切るよりは合理的かなと思います。
実際に動かすときは、以下のようにoptionを指定します。
out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen=c(240,160),
maxN=0, maxEE=c(2,2), truncQ=2, rm.phix=TRUE,
compress=TRUE, multithread=TRUE) # On Windows set multithread=FALSEPaird-end の場合は、長さが2のベクトルを与えてやります。そうすると、1個目がフォワード,2個目がリバースに対しての指定の値になります。
もし、フィルタリングの閾値がきつすぎて、使える read の値が0になったら、次の段階でエラーが出ます。
その場合は、maxEEの値を大きくしたり、truncQの値を下げたりして緩めてください。
もしくは、 read の値が0になったファイルを除く手もあります。僕は、精度の悪いデータを使いたくないので、除いています。
除き方は、Single-end の場合は
out <- filterAndTrim(fnFs, filtFs, truncLen=240,
maxN=0, maxEE=2, truncQ=2, rm.phix=TRUE,
compress=TRUE, multithread=TRUE)
# out は2列の matrix で、1列目にフィルタリング前のread数、2列目にフィルタリング後のread数が入っています
read0 <- which( out[,2]==0 )
filtFs <- filtFs[-read0]
Paird-end の場合は、out の中にフォワードもリバースも入っているので
out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen=c(240,160),
maxN=0, maxEE=c(2,2), truncQ=2, rm.phix=TRUE,
compress=TRUE, multithread=TRUE)
outF <- out[ grep( 'R1', rownams(out)), ] # フォワード側のファイル名が入った行を取り出す
read0F <- which( outF[,2]==0 )
filtFs <- filtFs[-read0F]
outR <- out[ grep( 'R2', rownams(out)), ] # リバース側のファイル名が入った行を取り出す
read0R <- which( outR[,2]==0 )
filtRs <- filtRs[-read0R]
といった、一工夫をしてあげてください。
長くなりましたが、これでひとまず終わります。
次回は、Error rate の計算について調べて、解説してみます。